Command Line Workflow
For these analyses, we’ll be starting with an aligned, trimmed, and sorted BAM file, test.bam. Alignment can be done using a variety of methods, including minimap2 and bwa. For information on how to perform trimming and sorting, check out the iVar manual.
Once you’ve got your BAM file, you’ll just need the reference that you
used for the alignment (i.e. Hu-1 for SARS-CoV-2 samples, like
this).Since we’re generally going to be
working with many wastewater samples at the same time, it’s a good idea
to create folders to store each of the output files, using
mkdir variants_files depth_files demix_files, for example. From
there you can go ahead and run initial single nucleotide variant (SNV)
calling step, in which we calculate the frequency of each observed
mutation in the data.
This can be done using the command
freyja variants test.bam --variants variants_files/test.variants.tsv --depths depth_files/test.depth --ref NC_045512_Hu-1.fasta
Before we perform demixing, it’s a good idea to make sure our list of lineage barcodes and corresponding metadata is up to date. We can do this by running
freyja update
which will save these files in freyja’s data folder. This can be a
bit tricky to find in your conda environment, so if you want a local
copy (good to keep around in case you want to compare results with
past/future barcode libraries) you can also add in the --outdir
option, and specify the location where you want to put the barcode
library. This just needs to be done once (per session – new lineages are
being added every day).You can also check the overal coverage depth status
across amplicons using the following command:
freyja ampliconstat --primer primer.bed --input_depth depth_files/test.depth
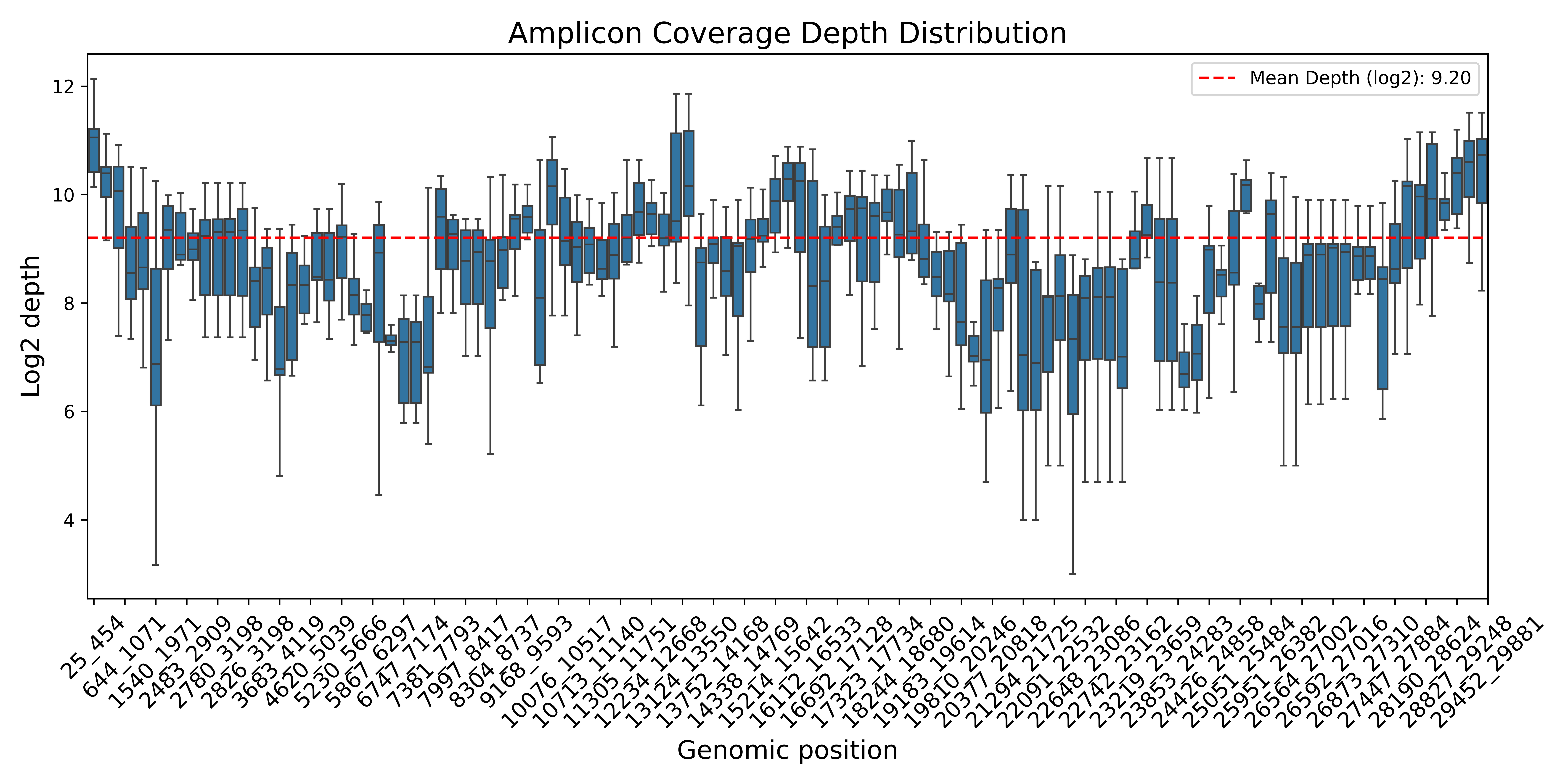
This command will creat a figure with distribution of depth for each
amplicon that is provided in the primer bed file. The plot will show genomic
coordinates of each amplicon, which can be cross-referenced with the
csv output created by this function. The csv file will provide
information such as amplicon number, genomic location,
mean coverage depth, amplicon length and amplification status.
The function use 5 as the threshold for minimum number of reads
present to discern the amplification status but you can set it
to a custom number using --min_depth.
Here is an example of how the plot will look like with the test data.
Then we can proceed to the de-mixing step. Demixing can be performed using the command
freyja demix variants_files/test.variants.tsv depth_files/test.depth --output demix_files/test.output --confirmedonly
By default, Freyja demix calculates the coverage across all genomic positions
in the provided reference genome and includes it in the output.
If you want to calculate the coverage in a specific genomic region, you can use
--region_of_interest and provide the regions in a JSON format following the
same structure as provided here
If you want to use your local barcodes set, you’ll need to use the
--barcodes option and specify the path of your local barcodes. Note:
While the output of freyja variants will not change over time, the
output of freyja demix depends strongly on the list of known lineage
barcodes. The method will assign the closest lineage (in an edit
distance sense) to what’s in the data, but if a more representative
lineage is identified the assignment will shift to the more
representative lineage.
Once you’ve run demix on a bunch of samples, you can aggregate all of the output files using the command
freyja aggregate demix_files/ --output bunch_of_files.tsv.
From there, it’s easy to look directly at the output files in any standard tsv viewer (Excel, Numbers, LibreOffice Calc, etc.).